Pathophysiology
I-4. Secondary effects of heart failure; therapeutic options
心不全の二次的影響と治療選択肢
Compensatory Mechanisms: Healthy vs HF
Short-term
- Frank-Starling law: ↑blood volume → stretch → stronger contraction. Particularly relied upon in HF.
- Sympathetic stimulation: ↑inotropy, chronotropy, dromotropy. HF patients gain little because their SYM system is already near maximum, so during exercise they depend on Frank-Starling.
Long-term (beneficial short-term, harmful long-term)
- Salt–water retention (RAAS): short-term ↑preload → ↑CO. Long-term → edema/pulmonary edema, ↑preload → heart works harder.
- Hypertrophy / remodeling.
Reaction to workload
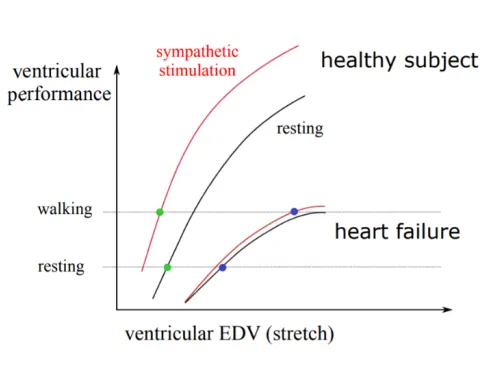
- Healthy: walking → SYM stimulation ↑contractility without raising EDV → plenty of CO reserve.
- HF: flat Frank-Starling curve, SYM has little effect → must use Frank-Starling → EDV rises on walking → very limited CO reserve.
Neurohormonal & Cellular Changes
Cellular
- Ca²⁺ leaks from SR via RyR2, SERCA activity ↓ → weaker contractions, impaired relaxation, longer contractions → prolonged QT → malignant arrhythmias.
- Energy shift: from fatty-acid oxidation to glucose oxidation, with fewer energy reserves → vulnerable heart, and ↓ATP/↑ADP → ↓lusitropy.
Neurohormonal response
- Hemodynamic defense reaction — short-term beneficial, long-term harmful:
| Mechanism | Short-term benefit | Long-term harm |
|---|---|---|
| Salt/water retention (↑preload) | ↑CO | Edema, pulmonary edema |
| Vasoconstriction (↑afterload) | ↑BP | ↓CO, ↑energy demand |
| Cardiac stimulation (↑contractility/HR) | ↑CO | ↑energy demand, arrhythmias, sudden cardiac death |
- Inflammatory reaction (low-grade): adaptive = protective proteins (heat shock), maladaptive = cardiac cachexia, apoptosis, necrosis.
- Hypertrophic response: cell stress + hemodynamic + inflammatory signals change gene expression → remodeling, ↑energy expenditure, ↑apoptosis → severe arrhythmias.
Treatment
Fundamentals: treat underlying cause, remove precipitating causes, symptomatic treatment, support heart & circulation (drugs + non-drug).
Symptomatic
- Diuretics: counteract RAAS-driven volume overload → ↓edema. Improve backward failure (↓preload) but can worsen forward failure (↓ejection/CO).
- ICD (implantable cardioverter-defibrillator): monitors rhythm. Paces bradyarrhythmia, terminates malignant arrhythmia via overdrive pacing or DC shock.
Drug therapy (“sick horse” analogy)
- ↑Contractility: digitalis (improves symptoms, no survival benefit, toxic — no longer used), β-agonists (improve symptoms but ↓survival, not for chronic use).
- ↓Load: vasodilators that block RAAS → improve symptoms and survival — ACE inhibitors, ARBs, aldosterone antagonists (block fibrosis).
- Slower, more effective pumping: β-blockers (may transiently worsen symptoms, improve long-term survival via RAAS blockade), and I_f-inhibitor ivabradine (for those who can’t tolerate β-blockers, e.g. asthma).
- Heart transplantation.
- LVAD (left ventricular assist device) — bridge to transplant, now usable for months; new devices fit in the pericardial cavity.
- Signaling pathways / new approaches: NEP (neprilysin) inhibitors (↑BNP), ARNI (valsartan/sacubitril), SGLT2 inhibitors (prolong survival regardless of diabetes). The “Fantastic 4”: ARNI, β-blocker, MRA, SGLT2-inhibitor. Also PDE5 inhibition, microRNA/gene therapy, stem-cell therapy.
一問一答
▶What are the short-term compensatory mechanisms in heart failure?
The Frank-Starling law (↑volume → stretch → stronger contraction) and sympathetic stimulation (↑inotropy, chronotropy, dromotropy).
▶Why do heart failure patients rely on the Frank-Starling mechanism during exercise?
Their sympathetic system is already near maximal, so it adds little; they must use Frank-Starling, but their flat curve gives very limited cardiac reserve.
▶Why is salt/water retention beneficial short-term but harmful long-term in heart failure?
Short-term it raises preload → ↑CO; long-term it causes edema/pulmonary edema and makes the heart work harder.
▶What cellular calcium-handling changes weaken the failing heart?
Ca²⁺ leaks from the SR via RyR2 and SERCA activity falls → weaker contraction, impaired relaxation, and prolonged contractions → prolonged QT → malignant arrhythmias.
▶How does cardiac energy metabolism change in heart failure?
It shifts from fatty-acid oxidation to glucose oxidation with fewer reserves, and ↓ATP/↑ADP impairs lusitropy → a vulnerable heart.
▶What are the three arms of the hemodynamic defense reaction and their long-term harms?
Salt/water retention (↑preload → edema), vasoconstriction (↑afterload → ↓CO, ↑energy demand), and cardiac stimulation (↑contractility/HR → ↑energy demand, arrhythmias, sudden death).
▶How does the inflammatory response affect the failing heart?
Low-grade inflammation can be adaptive (protective heat-shock proteins) or maladaptive (cardiac cachexia, apoptosis, necrosis).
▶Why is the hypertrophic remodeling response ultimately harmful?
Stress/hemodynamic/inflammatory signals change gene expression → remodeling with ↑energy expenditure and ↑apoptosis → severe arrhythmias.
▶What are the fundamentals of heart failure treatment?
Treat the underlying cause, remove precipitating factors, provide symptomatic treatment, and support the heart and circulation (drug + non-drug).
▶How do diuretics help and potentially harm in heart failure?
They counteract RAAS-driven volume overload → ↓edema and improve backward failure (↓preload), but can worsen forward failure by reducing ejection/CO.
▶What is the role of an ICD in heart failure?
It monitors rhythm, paces bradyarrhythmias, and terminates malignant arrhythmias via overdrive pacing or DC shock.
▶Why are positive inotropes (digitalis, β-agonists) limited in chronic heart failure?
They improve symptoms but provide no survival benefit (digitalis is toxic; β-agonists reduce survival), so they are not used for chronic therapy.
▶Which load-reducing drugs improve both symptoms and survival in heart failure?
RAAS-blocking vasodilators: ACE inhibitors, ARBs, and aldosterone antagonists (which also block fibrosis).
▶Why are β-blockers beneficial in heart failure despite transient worsening?
They may transiently worsen symptoms but improve long-term survival by blocking the harmful chronic RAAS/sympathetic activation.
▶What is the "Fantastic 4" of modern heart failure therapy?
ARNI (valsartan/sacubitril), β-blocker, MRA (mineralocorticoid receptor antagonist), and SGLT2 inhibitor.
▶When is ivabradine used in heart failure?
As an I_f-channel inhibitor that slows heart rate, for patients who cannot tolerate β-blockers (e.g., asthma).
▶What is the mechanism of ARNI in heart failure?
Neprilysin (NEP) inhibition raises beneficial BNP, combined with an ARB (valsartan/sacubitril) to reduce load.
▶What is notable about SGLT2 inhibitors in heart failure?
They prolong survival regardless of whether the patient has diabetes.
▶What is an LVAD and its role in heart failure?
A left ventricular assist device used as a bridge to transplant (now usable for months); new devices fit within the pericardial cavity.
▶How does the Frank-Starling response differ between a healthy heart and a failing heart on walking?
Healthy: sympathetic stimulation ↑contractility without raising EDV, preserving CO reserve. HF: flat curve and minimal sympathetic effect force reliance on Frank-Starling, so EDV rises with very limited CO reserve.